

A síndrome de Leigh é uma doença neurológica grave que normalmente se manifesta durante o primeiro ano de vida. No entanto um pequeno número de indivíduos não demonstra quaisquer sintomas até à idade adulta, ou então estes agravam-se lentamente com o tempo.
Esta doença é caracterizada por um atraso e perda progressiva das capacidades mentais e motoras, levando normalmente à morte nos primeiros dois a três anos de vida, usualmente devido a falha respiratória.
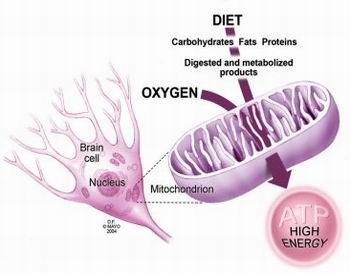
A mitocôndria é o principal produtor de energia, especialmente no sistema nervoso
Os primeiros sintomas da síndrome de Leigh consistem em vómitos, diarreia, e dificuldade em engolir, o que leva a uma menor ingestão de nutrientes. Isto resulta num crescimento lento, não se verificando o ganho de peso adequado à idade. Problemas musculares e motores são também comuns na síndrome de Leigh, entre os quais contrações musculares involuntárias e problemas de equilíbrio. Outra razão que leva à dificuldade de movimento é a perda de sensação e fraqueza nos membros inferiores.
Outras características da doença são ou a perda de movimento ocular ou movimentos oculares rápidos involuntários. Vários problemas respiratórios são também comuns em indivíduos com síndrome de Leigh.
Qual a taxa de incidência da síndrome de Leigh na população mundial?
A síndrome de Leigh afecta 1 em cada 40 000 recém nascidos, sendo no entanto mais comum em determinadas populações. Por exemplo, numa determinada região do Quebec, Canadá esta condição atinge aproximadamente 1 em cada 2000 recém nascidos. Nas Ilhas Faroé atinge 1 em cada 1700 recém nascidos.
Que alterações genéticas levam à ocorrência desta doença?
A síndrome de Leigh pode ser causada tanto por mutações no DNA nuclear ou no DNA mitocondrial, sendo que mutações no DNA mitocondrial ocorrem apenas em 20% dos casos.
A maioria dos genes afectados está envolvida no processo de produção de energia pela mitocôndria, a fosforilação oxidativa. Cinco complexos proteicos (complexo I, II, III, IV e V) estão envolvidos neste processo. Uma mutação que afete o seu funcionamento ou formação causa redução ou mesmo eliminação da sua atividade, o que leva à ocorrência de síndrome de Leigh.
Mutações em pelo menos um dos 25 genes (encontrados no DNA nuclear e no mitocondrial) envolvidos na formação do complexo I, são uma das principais causas da doença, sendo responsável por um terço dos casos.
Outra mutação, que afeta o complexo IV, também conhecido por citocromo c oxidase é responsável por 15% dos casos. Um dos genes mais frequentemente afetados que está na origem da síndrome de Leigh é o SURF1. Este gene, encontrado no DNA nuclear, codifica para uma proteína responsável pela “montagem” do complexo IV. Mutações neste gene levam tipicamente à formação de uma proteína anormal, que é degradada pelas células. A ausência de proteína funcional reduz a formação do complexo IV, reduzindo a produção de energia pela mitocôndria.
Relativamente a mutações no DNA mitocondrial, a mais comum na síndrome de Leigh ocorre no gene MT-ATP6. Este codifica para uma proteína que faz parte do complexo V, também denominado de ATP sintase, que utiliza a energia fornecida pelos outros complexos para gerar ATP, a principal forma de energia utilizada pela célula. Mutações neste gene afetam 10% dos indivíduos com síndrome de Leigh.
Para além destas, muitas outras mutações que diminuam a atividade dos vários complexos envolvidos na fosforilação oxidativa e consequentemente na produção de energia pela célula estão relacionadas com a síndrome de Leigh.
Como é a doença transmitida à descendência?

Diferentes padrões de hereditariedade da síndrome de Leigh. Crédito: NIH (adaptado)
A forma mais comum é por um padrão de hereditariedade autossomal recessivo, o que significa que ambas as cópias do gene têm de possuir mutação. Isto verifica-se nos genes presentes no DNA nuclear, como é o caso do SURF1. Neste caso os pais do indivíduo afetado possuem cada um uma cópia do gene mutado, não demonstrando tipicamente no entanto quaisquer sintomas.
Em 20% dos casos estamos perante um padrão de transmissão mitocondrial, também conhecido por hereditariedade materna. Uma vez que são os óvulos e não os espermatozoides que contribuem com as mitocôndrias para o desenvolvimento do embrião, a criança pode herdar doenças resultantes de mutações presentes no DNA mitocondrial da mãe.
É ainda possível ocorrer transmissão ligada ao cromossoma X, quando o gene mutado se localiza neste cromossoma, sendo mais comum em homens.
De que forma é feito o diagnóstico da doença?
É possível efetuar diagnóstico pré-natal caso a mutação se encontre no DNA nuclear. Nos casos em que o gene afetado se encontra no DNA mitocondrial, tal diagnóstico não é possível devido ao fenómeno de heteroplasmia (existência quer de mitocôndrias normais, quer de mitocôndrias com genoma alterado).
Qual o tratamento para a doença?
Não existe um tratamento específico para a doença. No entanto, administram-se vitaminas ou cofactores como a vitamina B1, B2 e coenzima Q cuja eficácia será dependente da causa subjacente da doença.
Fontes:
Raríssimas – Síndrome de Leigh