

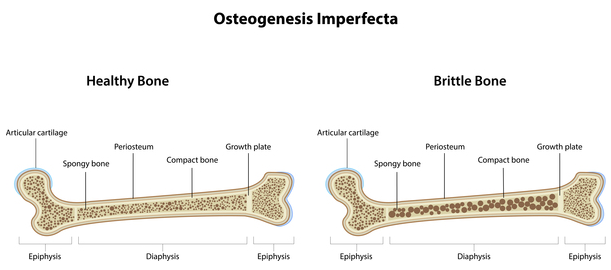
Esquema representativo da diferença entre um osso saudável e de um osso de um indivíduo com osteogenesis imperfecta
Osteogenesis imperfecta trata-se de um grupo de doenças genéticas que afetam principalmente a formação do osso, como sugerido pelo seu nome. Pessoas com esta doença possuem ossos frágeis, que sofrem fratura sem causa aparente. Múltiplas fraturas são comuns, ocorrendo mesmo antes do parto, nos casos mais extremos. Nos casos mais moderados podem apenas ocorrer algumas fraturas ao longo da vida do paciente, principalmente na infância e adolescência.
Existem pelo menos oito formas reconhecidas de osteogenesis imperfecta. Os diferentes tipos podem ser distinguidos consoante os sinais e sintomas que o paciente apresenta. O tipo I é a forma mais moderada de osteogenesis imperfecta, sendo o tipo II a forma mais agressiva. Todas as outras formas apresentam sinais que caiem algures entre as formas do tipo I e do tipo II.

Esclera azulada numa criança com osteogenesis imperfecta
Os pacientes com osteogenesis imperfecta do tipo I apresentam normalmente fraturas durante a infância e adolescência, normalmente sem grande impacto no seu estilo de vida. Estas fraturas tendem a ser menos recorrentes durante a fase adulta. As pessoas com esta forma da doença apresentam ainda uma esclerótica (parte branca do olho) mais azulada e podem perder a audição durante a fase adulta. Os indivíduos afetados apresentam estatura normal.
As formas mais graves da doença apresentam fraturas mais graves e frequentes que podem ocorrer mesmo antes do nascimento. Os indivíduos são normalmente de menor estatura, apresentam perda de audição desde a infância e possuem problemas respiratórios assim como um mau desenvolvimento da dentição (dentinogenesis imperfecta). A forma mais extrema de osteogenesis imperfecta, tipo II, inclui uma caixa torácica pequena e frágil assim como pulmões não desenvolvidos. Os recém-nascidos com estas características apresentam problemas graves, morrendo após o parto.
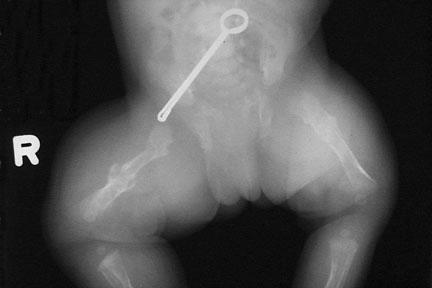
Fractura femural num recém nascido com osteogenesis imperfecta
A doença deve-se principalmente a mutações nos genes COL1A1 e COL1A2 que codificam para o colagénio do tipo I, constituinte da parte orgânica do osso e que lhe dá a sua flexibilidade.
A sua prevalência mundial é de 6 a 7 indivíduos afetados em cada 100 000 pessoas. Na maioria dos casos a doença trata-se de uma doença autossomal dominante, o que significa que basta um dos pais possuir o gene afetado para que a descendência apresente a doença. Pode também acontecer de novo, situação em que pais saudáveis geram um filho com a doença, ocorrendo a mutação durante o desenvolvimento embrionário.
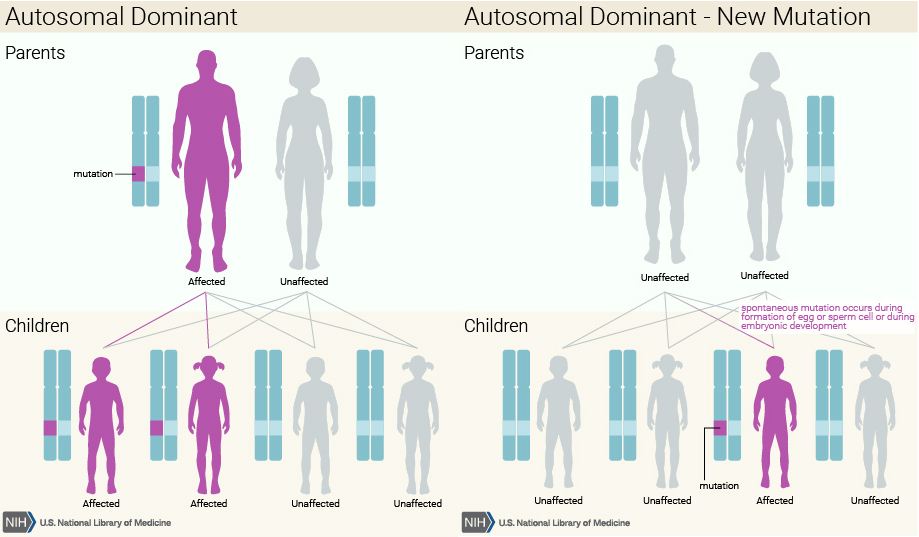
Diferença entre uma doença autossomal dominante com e sem mutação de novo
Fontes:
NIH – National Human Genome Research Institute