
O rearranjo de Fries, assim denominado em homenagem ao químico alemão Karl Theophil Fries, é uma reacção de rearranjo de um éster fenólico numa hidroxi-aril cetona catalisada por um ácido de Lewis, envolvendo a migração de um grupo acilo do éster fenólico para o anel arilo. Esta reacção é orto e para selectiva e pode favorecer-se a formação de um dos dois produtos possíveis ao alterar-se certas condições da reacção, tais como temperatura e solvente usado.
Mecanismo da reacção
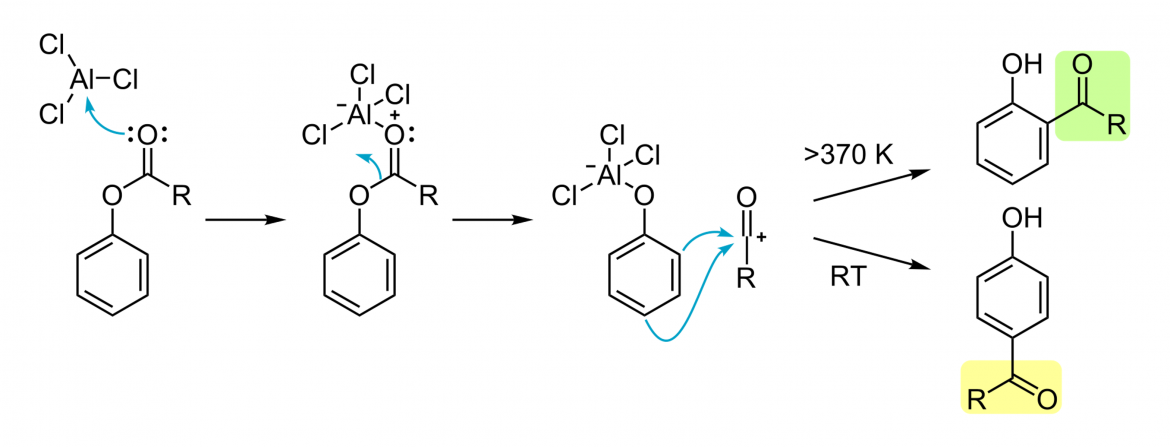
Apesar muito esforço, ainda não se possui uma imagem definitiva do mecanismo do rearranjo de Fries. Existe evidência que sustente mecanismo tanto inter- como intramolecular, obtidos por experiências de crossover com misturas de reagentes. O progresso da reacção não é dependente do solvente ou do substrato. Ainda assim, o mecanismo aceite envolve a formação de um intermediário carbocatião.
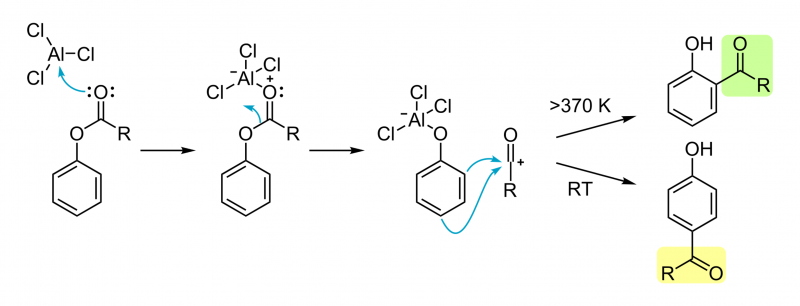
No primeiro passo da reacção, um ácido de Lewis (e.g. cloreto de Alumínio, AlCl3) coordena o oxigénio do carbonilo do grupo acilo. Este átomo de oxigénio é mais rico em densidade electrónica que o átomo de oxigénio fenólico, sendo por isso o ligando preferencial do cloreto de alumínio. Esta interacção polariza a ligação entre o grupo acilo e o átomo de oxigénio fenólico e o cloreto de alumínio rearranja-se para o oxigénio do grupo fenol. Isto gera um carbocatião acílio livre, o qual reage com o anel aromático via substituição aromática electrofílica clássica. O protão capturado é libertado sob a forma de ácido clorídrico, onde o cloreto é derivado do cloreto de alumínio. A orientação da reacção de substituição é dependente da temperatura: baixas temperaturas favorecem para-substituições, enquanto temperaturas elevadas favorecem a formação de produtos orto-substituídos. Este fenómeno é um exemplo clássico de controlo termodinâmico versus controlo cinético da reacção, já que o produto orto pode formar complexos bidentados, mais estáveis, com o átomo de alumínio. A formação do produto orto-substituído pode ser também favorecida em solventes apolares: à medida que a polaridade do solvente aumenta, a fracção de produto para-substituído aumenta.
Aplicações práticas
Fenóis originam ésteres em vez de hidroxi-aril cetonas quando reagem com halogenetos de arilo em condições de acilação de Friedel-Crafts. Assim sendo, o rearranjo de Fries tem relevância industrial no que diz respeito à síntese de hidroxi-aril cetonas, intermediários importantes indústria farmacêutica. Outros ácidos de Lewis que não o cloreto de alumínio podem ser usados para catalisar esta reacção, nomeadamente trifuoreto de boro, trofuorometanosulfunato de bismuto ou ácidos próticos fortes (e.g. fluoreto de hidrogénio ou ácido metanosulfónico).
Limitações
Em quaisquer circunstâncias, apenas se pode utilizar ésteres com componentes acilo estáveis que possam aguentar as condições severas do rearranjo de Fries. Se os componentes aromático ou acilo forem altamente substituídos, então o rendimento da reacção será reduzido, devido impedimento estérico. A desactivação de grupos meta-direccionados no grupo benzeno também tem efeitos adversos.
Rearranjo fotoquímico de Fries
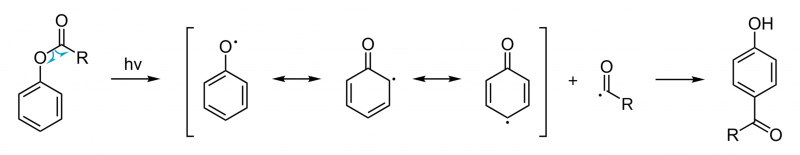
Além das condições normais da reacção térmica dos ésteres fenólicos, o rearranjo de Fries podem também ocorrer via processo fotoquímico. O rearranjo foto-Fries pode também originar produtos [1,3] e [1,5], envolvendo um mecanismo de reacção radicalar. Esta reacção é ainda possível com substituintes desactivadores no anel aromático. Uma vez que o rendimento é bastante baixo, este processo não é usado a nível comercial. No entanto, o rearranjo foto-Fries pode ocorrer naturalmente, por exemplo em objectos plásticos feitos de poliuretanos, poliésteres ou policarbonatos aromáticos quando expostos ao sol (os grupos carbonilo alifáticos sofrem reacções de Norrish, as quais são similares). Neste caso, a fotólise dos grupos éster leva à libertação de ftalatos dos plásticos.
Fonte: Wikipédia