
A atrofia muscular espinhal caracteriza-se por um grupo de distúrbios marcados pela degeneração progressiva dos neurónios motores na medula espinhal, resultando em fraqueza e atrofia muscular, geralmente sem evidência de lesão dos tratos corticoespinhais. As doenças nessa categoria incluem a doença de Werdnig-Hoffmann e, mais tarde, as ATROFIAS MUSCULARES DA INFÂNCIA ESPINHAL, a maioria das quais hereditárias.
O que é a AME?
A atrofia muscular espinhal (AME) é uma doença genética que afeta a parte do sistema nervoso que controla o movimento muscular voluntário.
A maioria das células nervosas que controlam os músculos estão localizadas na medula espinhal, o que explica a palavra espinhal no nome da doença. A AME é muscular porque o seu efeito primário ocorre nos músculos, que não recebem sinais dessas células nervosas. A atrofia é o termo médico para ficar menor, que é o que geralmente acontece com os músculos quando eles não estão ativos.
Para além disso, a AME envolve a perda de células nervosas chamadas neurónios motores na medula espinhal e é classificada como uma doença do neurónio motor.
Na forma mais comum da doença, há grande variabilidade na idade de início, sintomas e taxa de progressão. Para explicar essas diferenças, o cromossomo 5 AME é frequentemente classificado nos tipos 1 a 4.
A idade em que os sintomas da AME começam aproximadamente se correlaciona com o grau em que a função motora é afetada: Quanto mais cedo a idade de início, maior o impacto na função motora. As crianças que apresentam sintomas no nascimento ou na infância geralmente têm o nível mais baixo de funcionamento (tipo 1). AME com início em crianças (tipos 2 e 3), adolescentes ou adultos (tipo 4) e geralmente se correlaciona com níveis cada vez mais elevados de função motora.
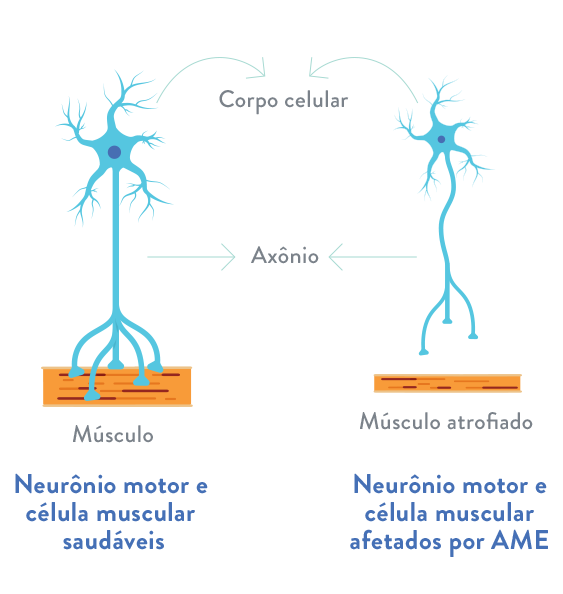
Figura 1 – Neurónio motor e célula msucular em indivíduo saudável vs. neurónio motor e célula muscular afetada pela doença AME.
Quais as causas do AME?
O cromossomo 5 AME é causado por uma deficiência de uma proteína do neurónio motor chamada SNM, para “sobrevivência do neurónio motor”. Essa proteína, como o próprio nome indica, parece ser necessária para a função normal do neurónio motor. Sua deficiência é causada por falhas genéticas (mutações) no cromossomo 5 em um gene chamado SNM1. Os genes vizinhos SMN2 podem em parte compensar os genes não funcionais do SNM1.
Outras formas raras de AME (não-cromossoma 5) são causadas por mutações em genes além da SNM.
Quais os sintomas de AME?
Os sintomas de AME podem variar de leve a grave. Veja o que se apresenta de seguida:
O principal sintoma da AME relacionada ao cromossomo 5 é a fraqueza dos músculos voluntários.
- Os músculos mais afetados são aqueles mais próximos do centro do corpo, como os dos ombros, quadris, coxas e parte superior das costas. Complicações especiais ocorrem quando os músculos usados para respiração e deglutição são afetados, resultando em anormalidades nessas funções. Se os músculos das costas se enfraquecerem, as curvaturas da coluna podem-se desenvolver.
Funcionamento sensorial, mental e emocional são inteiramente normais no cromossomo-5 AME.
Algumas formas de AME não estão ligadas à deficiência do cromossoma 5 ou SNM. Essas formas variam muito em gravidade e nos músculos mais afetados. Enquanto a maioria das formas, como a forma relacionada ao cromossomo 5, afeta principalmente os músculos proximais, existem outras formas que afetam principalmente os músculos distais (aqueles mais distantes do centro do corpo) – pelo menos no inicio da doença.
Qual a progressão da doença?
Na AME relacionada ao cromossomo 5, quanto mais tarde os sintomas começarem e quanto mais proteína SNM houver, mais soft será o curso da doença. Enquanto, no passado, os bebes que apresentasse a doença, normalmente não sobreviviam mais de dois anos, hoje a maioria dos médicos considera a AME relacionada à SNM como um contínuo e prefere não fazer previsões rígidas sobre a expectativa de vida ou fraqueza com base estritamente na idade de início da descoberta da doença.
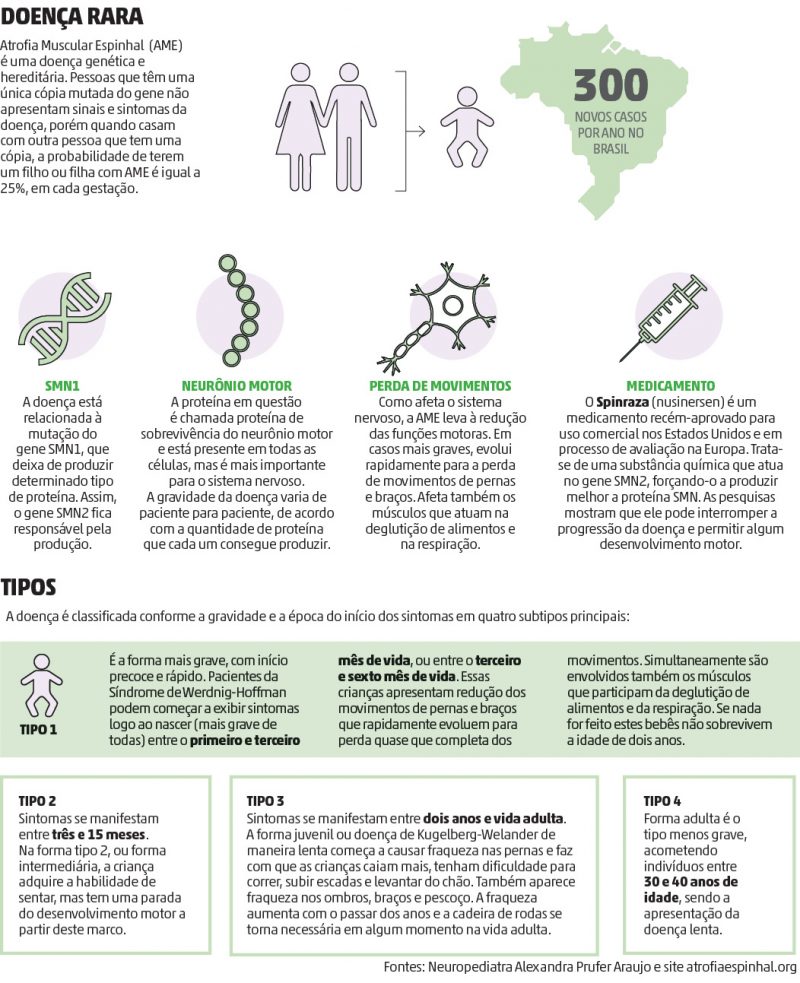
Figura 2 – Resumo da doença AME com algumas informações interessantes sobre o medicamento utilizado e os tipos de AME.
Fontes: MDA Muscular Dystrophy Association – Spinal Muscular Atrophy; ARTIGO: CELL Volume 80, Issue 1, 13 January 1995, Pages 155-165 – Identification and characterization of a spinal muscular atrophy-determining gene. Authors: SuzieLefebvre, LydieBürglen, SophieReboulle, OlivierClermont, PhilippeBurlet, LouisViollet, BernardBenichou, CorinneCruaud, PhilippeMillasseau, MassimoZeviani, DenisLe Paslier, JeanFrézal, DanielCohen, JeanWeissenbach, ArnoldMunnich, JudithMelki. ARTIGO: Nature Genetics volume 16, pages265–269 (1997) – Correlation between severity and SMN protein level in spinal muscular atrophy. Authors: Suzie Lefebvre, Philippe Burlet, Qing Liu, Solange Bertrandy, Olivier Clermont, Arnold Munnich, Gideon Dreyfuss e Judith Melki.