

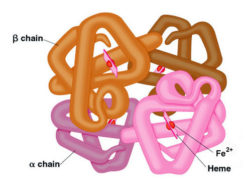
Estrutura da hemoglobina. Imagem retirada de The Medical Biochemistry Page
A hemoglobina é uma proteína presente nos glóbulos vermelhos sanguíneos, responsável pelo transporte de oxigénio desde os pulmões até todas as células do corpo. A constituição da hemoglobina é feita por 4 cadeias de péptidos, ligadas entre si: duas do tipo alfa e duas do tipo beta, gama ou delta, consoante o tipo de hemoglobina: A1 (a mais abundante), A2 e F (fetal).
As talassemias são um grupo de doenças que resultam da produção incorreta de cadeias alfa ou beta, por mutações genéticas. Esta produção anormal de hemoglobina traduz-se, por um lado, por uma menor afinidade desta para o oxigénio e, por outro, por uma diminuição na produção de hemoglobina e, portanto, de glóbulos vermelhos. Há também uma destruição acelerada de glóbulos vermelhos no baço e, até, dentro dos próprios vasos sanguíneos, levando a anemia.
As alfa-talassemias resultam de mutações nos genes que constituem as cadeias alfa da hemoglobina. Cada pessoa recebe de cada um dos pais, dois genes e, por isso, tem quatro genes que codificam para as cadeias alfa. Se apenas um gene estiver mutado, não há sinais nem sintomas de talassemia. Estes portadores, no entanto, podem passar a mutação para a geração seguinte. Se dois genes estiverem mutados, os sinais e sintomas serão ligeiros, constituindo assim uma alfa-talassemia minor. Três genes mutados resultam nos sinais e sintomas moderados a severos da doença da hemoglobina H. Quatro genes mutados dão origem à alfa-talassemia major, o que causa a morte do feto in utero ou à morte do bebé pouco após o nascimento (hydrops fetalis).
As beta-talassemias resultam de mutações nos genes envolvidos na produção das cadeias beta. Cada pessoa recebe um gene do pai e outro da mãe, tendo, no total, dois genes. Se um gene estiver mutado, há sintomas ligeiros, chamando-se assim, beta-talassemia minor. Dois genes mutados resultam na beta-talassemia major (também conhecida por anemia de Cooley), com sinais e sintomas moderados a severos. Os bebés com anemia de Cooley normalmente são saudáveis ao nascimento, mas começam a mostrar sintomas por volta dos dois anos. A mutação de dois genes pode também resultar numa beta-talassemia intermédia.
Sendo doenças genéticas, o risco de ter talassemia aumenta em pessoas com historial familiar de talassemias e, por isso, é importante fazer um diagnóstico pré-natal em casais de risco, a fim de evitar passar a doença para a geração seguinte. A talassemia é mais prevalente em pessoas com ancestralidade africana e do mediterrâneo.
Os sintomas das talassemias resultam da baixa capacidade da hemoglobina se ligar ao oxigénio, e por isso, de o fornecer a todas as células do organismo. Também há uma redução dos níveis de hemoglobina e da quantidade de glóbulos vermelhos no sangue. Assim, doentes com formas mais graves de talassemia terão sintomas mais severos, enquanto que os doentes com formas mais ligeiras têm sintomas mais leves, podendo até passarem despercebidos. Os sintomas incluem fadiga, fraquez e palidez, devido à anemia, icterícia (devido à rotura dos glóbulos vermelhos), e urina escura. Os doentes com talassemia desenvolvem também complicações, como deformidades nos ossos faciais, subdesenvolvimento nas crianças, esplenomegalia (aumento do baço) e problemas cardíacos.
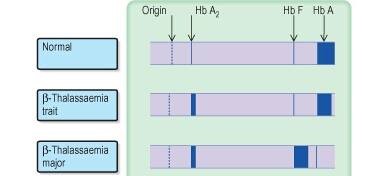
Eletroforese da hemoglobina. Imagem retirada de Medical Labs – Medical Laboratories Online.
O diagnóstico das talassemias é, normalmente, feito nas crianças, já que é em idades precoces que os sintomas começam a manifestar-se. O diagnóstico passa por uma análise sanguínea, que revela uma contagem de eritrócitos baixa, que serão de dimensões menores (microcíticos) e mais claros (hipocrómicos) do que o normal. Os níveis de hemoglobina são inferiores aos limites de referência (anemia), bem como os níveis de ferro. A eletroforese da hemoglobina permite saber que tipo de hemoglobina está em maior abundância, apontando para um possível diagnóstico. Uma análise ao DNA revelará se e quantos genes estão mutados, podendo definir com clareza que tipo de talassemia se trata.
O tratamento depende do tipo e da severidade da talassemia. As talassemias de baixa severidade não costumam requerer qualquer tipo de tratamento. Pode ser necessário, ocasionalmente, uma transfusão de sangue. As transfusões de sangue frequentes costumam ser o tratamento mais comum nos casos moderados a graves e, devido a este tratamento, algumas pessoas requerem terapia com quelantes de ferro para evitar o seu excesso. Os casos mais severos podem também requerer um transplante de medula com um dador compatível.
Hoffbrand AV, Moss PAH. Essential Haematology. 6th ed. Ltd JWS, editor2011.
Diseases and Conditions: Thalassemia. Mayo Foundation for Medical Education and Research (Consultado a 31-05-2016 em http://www.mayoclinic.org/diseases-conditions/thalassemia/basics/definition/con-20030316)