

Hoje trago um distúrbio do metabolismo de carboidratos designado de galactosemia. Descobre mais sobre esta temática com este post!
A galactosemia é causada por deficiências enzimáticas hereditárias que convertem a galactose em glicose. A sintomatologia inclui disfunções hepáticas e renais, deficiências cognitivas, cataratas e insuficiência ovariana prematura. O diagnóstico baseia-se na análise enzimática dos eritrócitos e do DNA. O tratamento é feito por eliminação da galactose da dieta. Com ele, o prognóstico físico é bom, mas os parâmetros cognitivos e de desempenho tornam-se frequentemente subnormais.
De referir que a galactose está presente nos laticínios, frutas e legumes. A transmissão das deficiências enzimáticas é autossómica recessiva.
Deficiência da galactose-1-fosfato uridil transferase
Esta deficiência causa a forma clássica da galactosemia.
A incidência é de 1/62.000 nascimentos e a frequência dos portadores é de 1/125.
Os lactentes normalmente são anoréticos e ictéricos com poucos dias ou semanas de amamentação com leite do peito ou uso de mamadeiras.
Para além disso, apresentam vómitos, hepatomegalia, crescimento deficiente, letargia, diarreia, anemia hemolítica, sepse (em geral por Escherichia coli) e, ainda, disfunção renal desencadeando acidose metabólica e edema.
Sem não houver tratamento nestes casos, a estatura das crianças permanece baixa e elas desenvolvem déficits cognitivos, da fala, do andar e equilíbrio durante a adolescência. Grande parte deles também desenvolvem cataratas, osteomalácia (causada por hipercalciúria) e insuficiência ovariana prematura.
Pacientes que apresentam a variante Duarte têm um fenótipo muito mais leve e a sintomatologia é claramente mais atenuada.
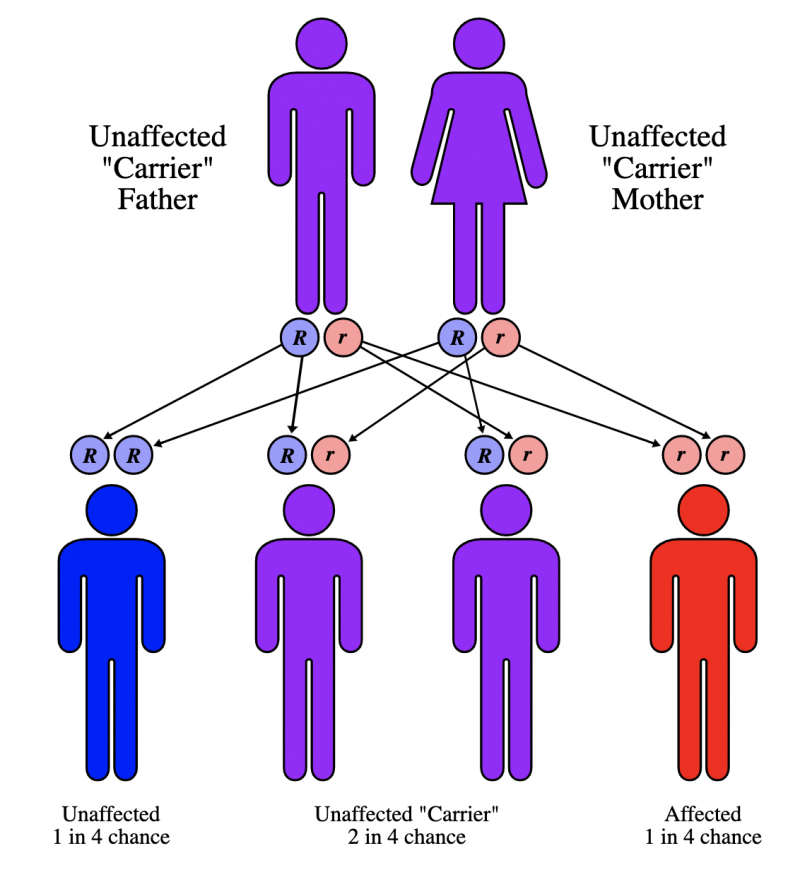
Figura 1 – Probabilidade de transmissão às gerações futuras.
Deficiência de galactoquinase
Os pacientes desenvolvem cataratas a partir da produção de galactitol, que lesa osmoticamente as fibras do cristalino, e raramente causa hipertensão intracraniana idiopática (pseudotumor cerebral).
A incidência é de 1/40.000 nascimentos.
Deficiência de galactose 4-epimerase uridina difosfato
Há fenótipos benignos e graves.
A forma benigna está restrita aos eritrócitos e leucócitos e não causa anormalidades clínicas. A forma grave causa uma síndrome muito semelhante à da galactosemia clássica, embora, às vezes, com perda da audição.
A incidência da forma benigna é 1/23.000 nascimentos no Japão; não há dados disponíveis sobre a incidência da forma mais grave.
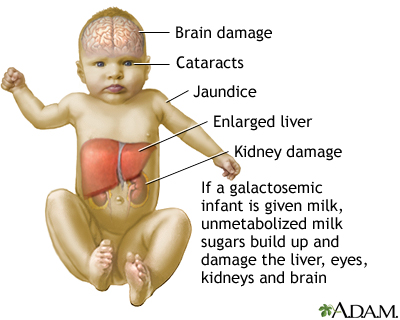
Figura 2 – Sintomatologia geral da galactosemia.
Diagnóstico da galactosemia
O diagnóstico baseia-se na avaliação dos níveis de galactose e por uma análise enzimática.
A presença de galactosemia é comprovada por níveis de galactose elevados e pela presença da redução em outras substâncias além da glicose (p. ex., galactose, galactose-1-fosfato) na urina.
Para além disso, é confirmado por análise de DNA ou pela análise enzimática dos eritrócitos, tecido hepático, ou ambos.
Tratamento da galactosemia
O tratamento da galactosemia baseia-se na restrição de galactose na dieta.
O tratamento da galactosemia consiste na eliminação na dieta de todas as fontes de galactose, mais notadamente a lactose (uma fonte de galactose), que está presente no leite materno, todos os produtos laticínios, incluindo leite artificial e é um adoçante usado em muitos alimentos.
A dieta livre de lactose previne a toxicidade aguda e reverte algumas manifestações mas pode não impedir deficiências neurocognitivas.