
A Doença de Fabry é também designada por doença de Anderson-Fabry, sendo considerada uma doença hereditária extremamente rara, até porque a transmissão ocorre de forma recessiva pelo cromossoma X, o cromossoma sexual feminino. Esta patologia caracteriza-se pela deficiência de uma enzima conhecida como alfa-galactosidase A.
Genética da doença
Normalmente os seres humanos apresentam 2 cromossomas sexuais, em que os homens apresentam uma cromossoma sexual Y e um cromossoma sexual X e as mulheres apresentam dois cromossomas sexuais X. A doença de Fabry é, portanto, uma doença que é transmitida pelo cromossoma X anómalo. Ao ser considerada uma doença com transmissão recessiva pelo cromossoma X, isto significa que para a doença se manifestar, no caso das mulheres, estas precisam de receber os 2 cromossomas X com defeito. A mulher só desenvolve doença nesta situação. No caso dos homens, estes só apresentam um cromossoma X e, portanto, basta que este cromossoma apresenta a anomalia para desenvolver a doença.
Desta forma, fica fácil perceber que a doença de Fabry é uma patologia que acomete mais os homens comparativamente com as mulheres e perceber que a doença nos homens é sempre herdada da mãe.
Assim, pode-se referir as seguintes características principais da doença:
- Ocorre fundamentalmente nos homens;
- Para uma mulher desenvolver a doença, precisa que ambos os pais (pai e mãe) tenham o cromossoma X anómalo;
- As mulheres costumam ser portadoras assintomáticas do cromossoma X anómalo, o segundo cromossoma X “protege-a” de desenvolver a doença;
- As mulheres portadoras assintomáticas do cromossoma X anómalo transmitem esse defeito para os descendentes.
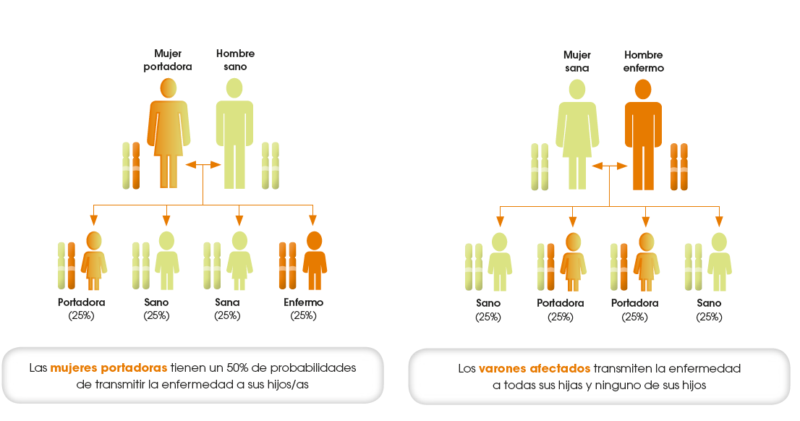
Figura 1 – Transmissão da doença
Causas da doença
A doença ocorre por uma anomalia na enzima alfa-galactosidase A (alfa-gal A) que é uma enzima presente dentro das células humanas e permite eliminar a globotriaosilceramida (GB3 ou GL-3) – substância lipídica. Assim, na ausência da alfa-gal A, a GB3 acumula-se dentro das células e deposita-se em vários tecidos, essencialmente ao nível da parede dos vasos sanguíneos. Desta forma, esta doença constitui um grupo de patologias chamado doenças de depósito.
Os sinais e sintomas desenvolvidos pelos portadores desta doença são derivados da acumulação de GB3 que obstrui os vasos sanguíneos e causa isquemias e enfarte nos tecidos e órgãos onde se verifique essa deposição.
Sintomas da doença
Os primeiros sintomas da doença, normalmente surgem logo na infância ou início da adolescência e incluem:
- Dores nos membros, principalmente mãos e pés, provocadas pelo acometimento dos nervos dos sistema nervoso periférico. Este é o principal sintoma e, muitas das vezes, é desencadeado pelo stress, frio, calor ou atividades físicas extenuantes.
- Lesões na pele – teleangiectasias – pequenos vasos da pele dilatados, também chamados de aranhas vasvulares. Comuns em pessoas com varizes nas pernas.
- Angioqueratomas são relevos puntiformes arroxeadas na pele.
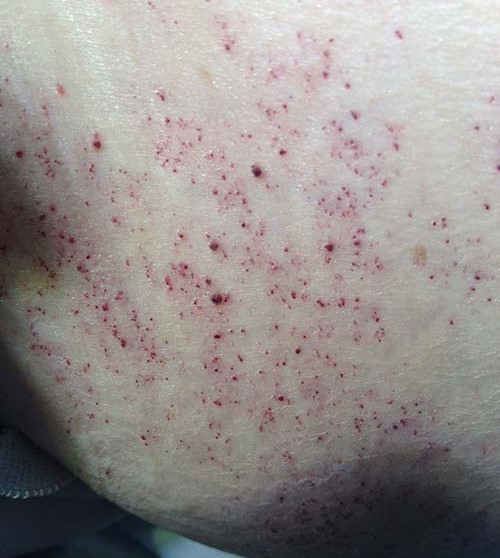
Figura 2 – Angioqueratomas
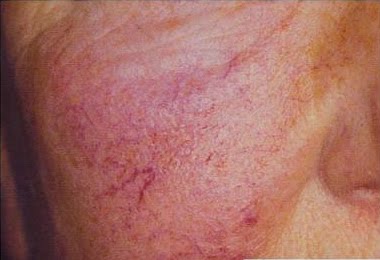
Figura 3 – Teleangieactasias (rosáceas)
Na doença de Fabry, as duas lesões costumam aparecer acopladas umas às outras, principalmente na zona das coxas, quadril e umbigo.
Outra das consequências é o comprometimento das glândulas sudoríparas em que os doentes apresentam baixa taxa de transpiração (hipoidorose), elevada temperatura corporal, fadiga intensa e intolerância ao calor.
Relativamente aos depósitos de GB3 nos vasos sanguíneos da córnea nos olhos e causa a córnea verticilata e, facilmente é detetada em exames oftalmológicos especiais.
Sintomas grastrointestinais também são comuns nesta patologia e, na idade adulta, a gravidade da doença é notória, quando os pacientes começam a sofrer obstruções nos vasos sanguíneos dos principais órgãos humanos, rins, cérebro e coração.
Diagnóstico da doença
O diagnóstico muitas das vezes é incerto e decorrer vários anos até um diagnóstico assertivo, isto porque a doença é rara e pouco conhecido, mesmo no âmbito clínico. tríade de alterações renais, angioqueratomas e dor nos membros em crianças ou jovens do sexo masculino deve, na maioria das vezes, levantar a suspeita de doença de Fabry.
O diagnóstico, quando incerto só pela observação da sintomatologia, pode ser confirmado através da dosagem da alfa-gal A, sendo que, a maioria dos pacientes apresentam níveis indetetáveis ou muito reduzidos desta enzima.
A biópsia à pele ou rimo porde fomentar o diagnóstico, nomeadamente nos casos atípicos da doença, em que o clínico não pensa inicialmente na probabilidade da doença de Fabry ser o diagnóstico final do doente.
As mulheres portadoras do gene podem ser assintomáticas e, portanto, apresentarem nível de alfa-gal a normais. Nestes casos, existe a encessidade de realizar testes genéticos para confirmar se a doente possui o gene da doença, podendo esta transmitir a doença aos seus descendentes.
Tratamento da doença
A doença de Fabry não tem cura.
Existe uma forma sintética da enzima alfa-gal A para administração permitindo tentar regular os seus níveis. A esperança média de vida dos portadores desta patologia ronda os 50 anos. Os medicamentos que existem para tratamento desta patologia são extremamente recentes e ainda não houve tempo para serem comprovados, em termos na diminuição da taxa de mortalidade, apesar das evidências notórias na redução dos depósitos de GB3. Quando mais precocemente for iniciado este tratamento, melhores serão os resultados clínicos obtidos com os pacientes. Este tratamento é par a vida toda!
Fontes: MD.Saúde – Doença de Fabry, Portal da diálise – Doença de Fabry, ABRAFF – Doença de Fabry